寡核苷酸修饰鬼臼毒素类衍生物及其制备方法和应用
文献发布时间:2024-04-18 19:52:40
技术领域
本发明属于鬼臼毒素类衍生物领域,具体涉及寡核苷酸修饰鬼臼毒素类衍生物及其制备方法,本发明还涉及所述寡核苷酸修饰鬼臼毒素类衍生物在制备抗肿瘤药物中的用途。
背景技术
鬼臼毒素(podophyllotoxin)是一种从天然盾叶鬼臼中分离提取得到的,具有抗病毒和抗肿瘤活性的木脂素类化合物,鬼臼毒素(Podophyllotoxin)结构如图19中式(Ⅰ)所示,研究学者致力于对鬼臼毒素母核进行有效的修饰,以得到保持原有或甚至更高的抗肿瘤活性,且毒副作用较小的鬼臼类衍生物。例如,在其基础上C环4位以氮杂环取代获得一种衍生物,为5-氨基-吲唑-鬼臼毒素,进一步提高了鬼臼毒素的抗肿瘤活性,但因强烈的毒副作用和生物利用度差等缺点,限制了它们在临床的应用。
发明内容
本发明的目的之一是提供一类具有良好安全性的寡核苷酸修饰鬼臼毒素类衍生物;
本发明的目的之二是提供一种制备所述寡核苷酸修饰氮杂环取代鬼臼毒素类衍生物的方法;
本发明的目的之三是将所述寡核苷酸修饰氮杂环取代鬼臼毒素类衍生物应用于制备临床用抗肿瘤药物。
本发明的上述目的是通过以下技术方案来实现的:
一类具有抗肿瘤活性的寡核苷酸修饰氮杂环取代鬼臼毒素类衍生物或其药学上可接受的盐,所述鬼臼毒素类衍生物的结构式为(Ⅱ)所示:
其中,R
其中,*为这些结构与氮原子的附接点;
R
R3为寡核苷酸序列。
本发明采用的寡核苷酸是核苷酸序列确定的,具有良好水溶性的柔性链状化合物,能够靶向肿瘤微环境中的半乳糖凝集素1,序列为AP-74M545,以其作为修饰基团有利于提高鬼臼毒素衍生物的水溶性以及靶向性,通过靶向肿瘤组织提高鬼臼毒素衍生物的安全性。
核苷酸序列AP-74M545如SEQ ID No.1所示。
本发明提供的制备上述式(Ⅱ)所示化合物的方法,包括以下步骤:
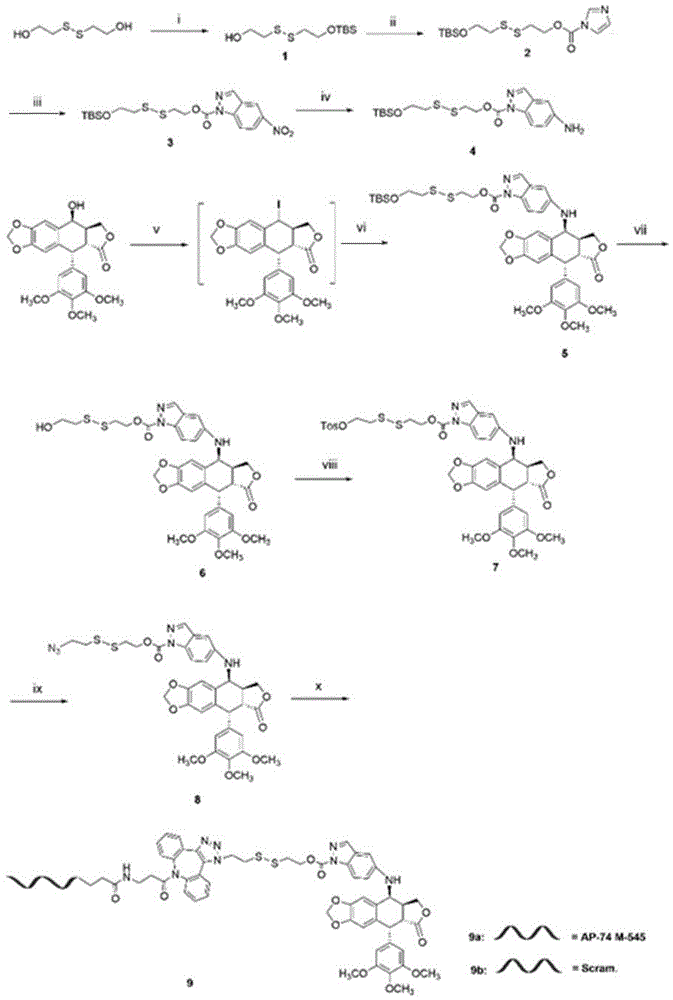
步骤(1),利用TBS基团保护2-羟乙基二硫化物的一羟基端,得到化合物1:
步骤(2),通过缩合反应,将N,N-碳酰二咪唑的N-甲酰基咪唑连接到化合物1的羟基,得到化合物2:
步骤(3),在有机溶剂三中,加入有机碱催化剂,用反应单体的取代化合物2的咪唑,获得化合物3,有机碱为1,8-二氮杂双环[5.4.0]十一碳-7-烯和4-二甲氨基吡啶,有机溶剂为乙腈;反应单体选自5-硝基-吲唑、6-硝基-吲唑、7-硝基-吲唑、5-硝基-吲哚、6-硝基-吲哚、7-硝基-吲哚中的一种,化合物3为
中的一种;
步骤(4),利用加氢反应,将化合物3的硝基转化为氨基,得到化合物4,使用的催化剂一为钯碳;
步骤(5),通过碘取代反应,在鬼臼毒素的C环4位引入碘,获得碘代鬼臼毒素;通过亲和取代反应将化合物4的引入碘代鬼臼毒素的C环4位,获得化合物5;
步骤(6),用HCl去除化合物5的TBS基团,暴露出自由的羟基,获得化合物6;
步骤(7),将化合物6溶解于吡啶中,加入对甲苯磺酰氯反应后,使对甲苯磺酰基取代化合物6的羟基,获得化合物7;
步骤(8),将化合物7溶解于N,N-二甲基乙酰胺,加入叠氮化钠,使叠氮基取代对甲苯磺酰基,获得化合物8;
步骤(9),将化合物8与5’端被DBCO修饰的寡核苷酸溶解在双相溶液中,进行无铜的点击化学反应获得化合物9即式(Ⅱ)所示的化合物,其中双相溶液为磷酸盐缓冲液和二甲基亚砜的等体积混合物。
共轭物的合成需要在硫桥的两端利用寡核苷酸和氮杂环鬼臼毒素类衍生物进行修饰,为了增加产量,核苷酸的添加被放在合成的最后一步进行。因此,合成开始时,在咪唑存在的情况下,用TBS基团保护硫桥上的一端羟基,得到化合物1;化合物2由CDI取代化合物1另一端的羟基得到;化合物3用同样的方法将化合物2与5-硝基-吲唑模块取代生成;为了使化合物3以C-NH键的形式与碘代鬼臼毒素连接,化合物3中的硝基被Pd/C还原成氨基,得到化合物4,将化合物4在TEA和BaCO
本发明中,所述反应步骤可以在以下条件下进行:(1)将2-羟乙基二硫化物溶于有机试剂,随后加入咪唑,混匀后加入叔丁基二甲基氯硅烷。溶剂可为四氢呋喃等;(2)将上一步反应产物溶于有机试剂,加入N,N-碳酰二咪唑,在常温下搅拌。溶剂可为二氯甲烷等;(3)将5-硝基-吲唑和上一步反应产物溶于有机溶剂中,加入有机碱催化剂,在常温下搅拌,有机碱可以为1,8-二氮杂双环[5.4.0]十一碳-7-烯和4-二甲氨基吡啶,有机溶剂可以为乙腈等,(4)在甲醇存在的条件下,将上一步产物混合钯碳,在氢气的氛围下进行硝基的还原,(5.1)鬼臼毒素溶于有机试剂中,加入碘化钠,有机试剂可以为三氟化硼乙醚和乙腈,(5.2)将上两步的产物混合于有机溶剂中,加入催化剂,有机试剂可以为三氟乙酸,四氢呋喃等,催化剂为碳酸钡,缚酸剂为三乙胺或吡啶;(6)将上步反应产物置于溶剂中,搅拌过夜,溶剂为体积比为1的1MHCl/THF,(7)将上步反应产物溶解于有机溶剂中,加入对甲苯磺酰氯,有机溶剂可以为吡啶,(8)将上一步产物溶解于N,N-二甲基乙酰胺,加入叠氮化钠后搅拌4个小时,(9)将上一步产物与DBCO修饰的寡核苷酸溶解在双相溶液中,进行无铜的点击化学反应,其中双相溶液为磷酸盐缓冲液和二甲基亚砜的等体积混合物。
本发明中,在所有反应中,各反应单体之间的摩尔比可以为1:1~10。
本发明中,所述所有反应温度可以为-20~50℃。
进一步的,本发明中,所述化合物1在0℃下制备获得,其他化合物制备过程的反应温度为常温。
上述制备方法还包括:将步骤(1)、步骤(2)、步骤(3)反应后的反应液倾入20~50倍体积的去离子水中,用二氯甲烷萃取,干燥后得化合物2、化合物3、化合物4的粗产物。
进一步,还包括将所述粗产物依次使用硅胶柱层析和凝胶柱层析分离,即得纯化的氮取代鬼臼毒素类衍生物产物。
优选的,所述硅胶柱层析分离方法包括:(1)硅胶柱层析为正相硅胶柱层析或反相硅胶柱层析,正相硅胶以低极性有机溶剂拌匀后装柱,以洗脱剂平衡,所述的洗脱剂优选是体积比为20:1的石油醚和乙酸乙酯组成;反相硅胶以甲醇拌匀后装柱,以洗脱剂平衡,所述的洗脱剂优选是体积比为60:1的甲醇和水组成;(2)将待分离纯化的样品以洗脱液溶解,进行上样吸附,然后用洗脱剂洗脱,收集洗脱液,将样品挥干并重结晶;
体外HepG2细胞活性抑制的测试表明,本发明制备的式(Ⅱ)所示化合物的抗肿瘤活性与5-氨基-吲唑-鬼臼毒素的抗肿瘤活性基本相当;选取活性最好的5-氨基-吲唑-鬼臼毒素-寡核苷酸共轭物(AP74-IZP)进行增殖抑制的测试表明,本发明制备的式(Ⅱ)所示化合物具有优于5-氨基-吲唑-鬼臼毒素和上市药物依托泊苷、奥沙利铂的抗增殖能力。体外微量热涌动测试表明,本发明植被的式(Ⅱ)所示化合物能够靶向寡核苷酸的受体蛋白半乳糖凝集素-1。体内HepG-2皮下移植瘤试验结果表明,本发明式(Ⅱ)所示化合物具有优于5-氨基-吲唑-鬼臼毒素和上市药物依托泊苷、奥沙利铂的体内抗肿瘤活性。体内Hepa1-6皮下移植瘤试验结果表明,本发明式(Ⅱ)所示化合物具有优于5-氨基-吲唑-鬼臼毒素和上市药物依托泊苷的体内抗肿瘤活性,并表现出逆转T细胞耗竭,产生免疫治疗效果的能力,以上测试结果证明本发明式(Ⅱ)所示化合物可制备成抗肿瘤药物,临床应用于抗肿瘤治疗。
本发明提供的抗肿瘤药物组合物,包括有效量的式(Ⅱ)所示的化合物或其盐和药学上可接受的载体,即将药学上可接受用量的式(Ⅱ)化合物与药学上可接受的载体配合后,按本领域常规的制剂方法将其制备成任意一类适宜的药物组合物。通常该药物组合物适合于口服给药和注射给药,也适合其他的给药方法,例如经皮给药。该药物组合物可以是片剂、胶囊、粉剂、颗粒、锭剂、栓剂,或口服液或无菌胃肠外悬液等液体制剂形式。该组合物可以是大或小容量注射剂、冻干粉针、无菌粉分装等制剂形式。为了达到给药的一致性,本发明药物组合物优选为单剂形式。用于口服给药的单剂形式可以是片剂和胶囊,并可含有常规赋形剂诸如粘合剂,例如糖浆、阿拉伯胶、明胶、山梨醇、黄芪胶或聚乙烯吡咯烷酮;填充剂,例如乳糖、糖、玉米淀粉、磷酸钙、山梨醇或甘氨酸;压片润滑剂,例如硬脂酸镁;崩解剂,例如淀粉、聚乙烯吡咯烷酮、淀粉乙醇酸钠或微晶纤维素,或药学上可接受的湿润剂,诸如十二烷基硫酸钠。
附图说明
图1为化合物9对HepG2细胞的克隆形成;
图2为化合物9对HepG2细胞的周期阻滞;
图3为化合物9的小动物体内分布(各脏器);
图4化合物9等对HepG2皮下移植瘤小鼠模型的治疗效果;
图5化合物9等对HepG2皮下移植瘤小鼠模型的治疗效果;
图6化合物9等对HepG2肿瘤给药后的H&E染色;
图7化合物9等对HepG2肿瘤给药后的Ki67-IHC;
图8化合物9等对HepG2肿瘤小鼠给药的体重变化;
图9化合物9等对HepG2肿瘤小鼠给药的脏器系数变化;图中每个脏器的柱状数据从左至右依次对应Nomalmice、Vehicle、AP74、Oxaliplatin、Etoposide、IZP、AP74-IZP;
图10为化合物9等对荷HepG2肿瘤小鼠给药的血象变化;
图11为化合物9等对HepG2肿瘤小鼠给药的血生化指标变化;
图12为化合物9等对HepG2肿瘤小鼠给药的器官损伤(H&E);
图13为化合物9等对Hepa1-6肿瘤C57小鼠的肿瘤体积缩小情况;
图14为化合物9等对Hepa1-6肿瘤C57小鼠的肿瘤质量缩小情况;
图15为化合物9等对Hepa1-6肿瘤组织内CD4/CD8变化的影响;
图16为化合物9等对Hepa1-6肿瘤组织内CD4/CD8变化的影响;
图17为化合物9等对Hepa1-6肿瘤组织内各细胞因子表达量变化的影响;
图18为化合物9的制备过程;
图19为5-氨基-吲唑-鬼臼毒素的分子式。
具体实施方式
下面通过具体实施例,对本发明的技术方案做进一步说明。
本发明所采用原料和设备若非特指,均可从市场购得或是本领域常用的,实施例中的方法,如无特别说明,均为本领域的常规方法。
试验材料
1、鬼臼毒素:均购自西安和霖生物工程有限公司;
2、5-硝基-吲唑、叔丁基二甲基氯硅烷、N,N-羰基二咪唑、咪唑、1,8-二氮杂双环[5.4.0]十一碳-7-烯、均购自上海毕得医药有限公司。
3、DBCO修饰的寡核苷酸合成自上海生物工程有限公司。
实施例15-氨基-吲唑-鬼臼毒素-寡核苷酸共轭物(化合物1)的合成和纯化
(1)5-氨基-吲唑-鬼臼毒素-寡核苷酸共轭物的合成:
合成中间体1
将10mmol二(羟乙基)二硫化物溶解于40ml四氢呋喃中,在0℃下搅拌,随后加入咪唑,加氮气保护,在室温条件下搅拌30min后,加入TBSCl(叔丁基二甲基氯硅烷),反应8h,用来保护二(羟乙基)二硫化物的其中一个羟基。以上合成反应平行实施4组,共计投料40mmol二(羟乙基)二硫化物。反应完成后200mLDCM分3次萃取,收集有机相,饱和NaCl水溶液分3次洗涤,再经无水Na
合成中间体2
取上述化合物1(27.9mmol)于250mL烧瓶中,加入100mL二氯甲烷溶解,在常温下加N,N-羰基二咪唑(41.8mmol,6.8g),磁力搅拌下3h。TLC检测反应后,200mL DCM分3次萃取,收集有机相,饱和NaCl水溶液分3次洗涤,再经无水Na
合成中间体3
取上述化合物2(3.32mmol)于25mL圆底烧瓶中,加入10mL乙腈溶解,在常温下加反应单体:5-硝基吲唑(4.93mmol),再加1,8-二氮杂二环十一碳-7-烯(DBU0.24mmol)。TLC检测反应后,收集所有反应液100mLDCM分3次萃取,收集有机相,饱和NaCl水溶液分3次洗涤,再经无水Na
合成中间体4
取上述化合物3600mg于25mL烧瓶中,加入5mL甲醇和催化剂钯碳,加橡胶塞密闭后用真空泵除尽空气,插上氢气球。TLC检测反应完全,抽滤后的滤液经无水Na
合成中间体5
取2mmol的PTOX(Podophyllotoxin,鬼臼毒素)于50ml圆底烧瓶,加入3.0mmol的NaI,加10ml的乙腈溶解,在冰浴下加入6.0mmol的BF
合成中间体6
将800mg化合物5溶于体积比1:1的四氢呋喃和HCl(2M)的混合溶剂中,在常温下搅拌。TCL检测反应结束,加300mL水,50mLEA(乙酸乙酯)分3次萃取,收集有机相,饱和NaCl水溶液分3次洗涤,经无水Na
合成中间体7
将化合物6200mg溶于4mL吡啶中,搅拌下加入对甲苯磺酰氯,TLC监测反应进展,直至反应完成,用EA(2×30mL)提取,有机相结合,用饱和NaCl水溶液洗涤,无水Na
合成中间体8
将化合物7100mg溶于3mLDMF中,在室温下搅拌加入叠氮化钠70mg,通常在过夜后TLC板监测,至反应完全。加水100mL,50mLEA分3次萃取,收集有机相,饱和NaCl水溶液分3次洗涤,经无水Na
合成产物9
将DBCO(二苯并环辛炔)修饰的寡核苷酸(20nmol)在0.8mL的20mM磷酸盐缓冲液(PBS)(pH7.2)中和化合物8(25nmol)在10mL烧瓶中混合。将混合溶液在室温搅拌过夜。然后取样液相分析反应进程。反应完全,通过三乙胺乙酸体系HPLC制备分离拿到终产物,即图18中的淡黄色粉末化合物9。
寡核苷酸序列AP-74M545,如SEQ ID No.1所示。SEQ ID NO:1:5′-DBCO-TTACAGTCGGCTGTAATTTAGTGTATGTACCGGTGTGTGTACGAT--3′。
(2)分离和纯化AP74-IZP,
使用硅胶柱层析和HPLC制备分离纯化。
(A)用正相硅胶柱(正相硅胶:中国青岛海洋化工有限公司,HG/T2354-92;分离系统:瑞士步琪(Buchi)等度快速色谱系统;色谱柱:瑞士步琪(Buchi)玻璃柱C-690,长460mm,内径15mm)或者相似极性柱分离;以上8种化合物分离纯化时洗脱液分别为(1)石油醚:乙酸乙酯=15:1。(3)石油醚:乙酸乙酯=30:1。(4)石油醚:乙酸乙酯=3:1。(5)石油醚:乙酸乙酯=3:1。(6)二氯甲烷:甲醇=150:1。(7)石油醚:乙酸乙酯=3:1。(8)石油醚:乙酸乙酯=3:1,以上8种化合物分离纯化时上样量2ml,流速恒流1.0ml/min;每2ml洗脱液作为一个馏分进行收集。用正相硅胶薄层(德国莫克高效硅胶薄层)或相似极性薄层检视各馏分;石油醚:乙酸乙酯3:1系统作为展开剂,将Rf值0.5的馏分进行合并;将合并后的样品真空干燥,避光条件下保存于4℃的冰箱中作为供纯化样品。
(B)用高效液相色谱(色谱柱:C18;长280mm,内径0.5mm)分离;以甲醇平衡,三乙胺乙酸:甲醇梯度洗脱条件为:将待纯化样品用双蒸水稀释,以0.6ml/min的流速上样吸附,然后梯度洗脱,目标产物在第10.2min出峰,每90μL洗脱5mL,洗脱液冻干得到终产物。
化合物12-(2-(叔丁基二甲基硅氧基)乙基)二硫酰基)乙炔-1-醇:无色油状液体,C
1
化合物22-((2-(叔丁基二甲基硅氧基)乙基)二硫基)乙基1H-咪唑-1-羧酸酯:淡黄色油状液体,C
1
化合物32-((2-(叔丁基二甲基硅氧基)乙基)二硫基)乙基5-硝基-1H-吲唑-1-羧酸酯:淡黄色蜡状固体,C
1
化合物42-((2-(叔丁基二甲基硅氧基)乙基)二硫基)乙基5-氨基-1H-吲唑-1-羧酸酯:淡黄色油状液体,C
1
化合物52-((2-(叔丁基二甲基硅氧基)乙基)二硫基)乙基5-氨基-1H-吲唑-鬼臼毒素:淡黄色固体,C
1HNMR(600MHz,CDCl3,298K):δ(ppm)=8.04(d,J=8.9Hz,1H),8.02(s,1H),6.87(dd,J=9.0,2.3Hz,1H),6.75(s,1H),6.50(s,1H),6.31(s,2H),5.93(dd,J=12.0,1.1Hz,2H),4.73(t,J=6.9Hz,2H),4.72–4.69(m,1H),4.57(d,J=4.9Hz,1H),4.41(t,J=8.0Hz,1H),4.13(d,J=5.8Hz,1H),3.97(dd,J=10.7,8.6Hz,1H),3.85(t,J=6.6Hz,2H),3.78(s,3H),3.73(s,6H),3.16(dd,J=14.1,5.0Hz,1H),3.11(t,J=6.9Hz,2H),3.08–3.00(m,1H),2.85(t,J=6.6Hz,2H),0.87(s,9H),0.05(s,6H).13CNMR(151MHz,CDCl3,298K):δ(ppm)=173.59,151.65,149.28,147.38,146.71,143.65,138.70,136.34,134.00,130.80,129.30,126.11,117.40,114.74,108.99,108.03,107.32,100.60,98.38,67.78,64.43,60.74,59.75,55.29,52.26,42.57,40.93,40.53,37.66,35.60,17.31,0.00,-6.26.ESI-ToF:[C40H49N3O3S2Si+H]+846.4382(CalculatedforC
化合物62-((乙基)二硫基)乙基5-氨基-1H-吲唑-鬼臼毒素:淡黄色固体,C
1
化合物72-(2-甲苯磺酰氧基)乙基二硫基乙基-5-氨基-1H-吲唑-鬼臼毒素:淡黄色固体,C
1HNMR(600MHz,CDCl3,298K):δ(ppm)=7.91(s,1H),7.34(d,J=8.8Hz,1H),6.78(s,1H),6.77(dd,J=8.9,2.1Hz,1H),6.71(d,J=1.5Hz,1H),6.53(s,1H),6.35(s,2H),5.95(d,J=11.3Hz,2H),4.70(d,J=4.0Hz,1H),4.60(d,J=5.0Hz,1H),4.44(t,J=8.0Hz,1H),4.04(dd,J=10.6,8.7Hz,1H),3.82(s,3H),3.76(s,6H),3.22(dd,J=14.0,5.0Hz,1H),3.09–3.01(m,1H).13CNMR(151MHz,CDCl3,298K):δ(ppm)=173.74,151.57,149.20,147.21,146.56,144.15,143.89,138.92,136.16,134.17,131.61,130.70,129.38,128.96,126.90,126.15,117.47,114.55,108.83,108.16,107.26,100.55,98.33,67.84,66.67,64.04,59.70,55.22,51.98,42.55,41.25,40.84,37.67,35.79,20.63.
化合物82-(叠氮乙基)二硫基乙基-5-氨基-1H-吲唑-鬼臼毒素:淡黄色固体,C
1
化合物95-氨基-吲唑-鬼臼毒素-寡核苷酸共轭物(AP74-IZP):淡黄色粉末。
[M]
试验例1本发明实施例制备的化合物抑制肿瘤细胞的活性试验
一、试验材料
1、供试化合物:实施例化合物9
2、对照化合物:5-氨基吲唑-鬼臼毒素,系本实验室合成,纯度98%;依托泊苷(购自上海毕得医药);奥沙利铂(购自上海毕得医药);
3、细胞株:HepG2、Huh7、PLC/PRF/5和SNU387细胞株购自武汉普诺赛生物科技有限公司;
二、试验方法
将对数生长期的HepG2、Huh7、PLC/PRF/5和SNU387细胞株,1000rpm离心5分钟,弃上清,适量培养基悬浮,调整细胞浓度至7000-10000个/孔,将细胞接种于96孔培养板中,设置以下实验组别:
1个阴性对照组(1‰DMSO,即溶剂对照组);试验组(即化合物9);3个对照组:5-氨基吲唑-鬼臼毒素组、依托泊苷组和奥沙利铂组。
每孔0.10mL细胞,用含有10%小牛血清的RPMI1640作为培养液,37℃、5%CO
三、试验结果
试验结果见表1,由表1可知,化合物9对HepG2、Huh7、PLC/PRF/5和SNU387细胞株的抗肿瘤活性较5-氨基吲唑-鬼臼毒素以及目前已做为抗肿瘤药物上市的鬼臼毒素类衍生物依托泊苷和抗肝癌上市药物奥沙利铂的抗肿瘤活性均有明显提高;且水溶性明显较好。
表1寡核苷酸取代5-氨基吲唑-鬼臼毒素对体外肿瘤株的IC
经活性评价后,筛选出HepG2细胞作为活性最好的细胞,进行了细胞克隆形成实验。
由图1可知,化合物9与5-氨基吲唑-鬼臼毒素展现出相同的抗细胞增殖能力,细胞克隆团明显减少,抗增殖能力较依托泊苷和奥沙利铂有明显提高。
经抗增殖能力评价后,对化合物9和5-氨基吲唑-鬼臼毒素、依托泊苷和奥沙利铂进行了细胞周期阻滞实验。
由图2可知,化合物9展现出与5-氨基吲唑-鬼臼毒素相近的周期阻滞能力,能将HepG2细胞几乎完全阻滞在G2/M期,明显强于依托泊苷和奥沙利铂。
经周期阻滞效果评价后,对化合物9进行了体内分布实验。
由图3可知,化合物9主要分布在肝、肾和肿瘤部位,相较于半乳糖凝集素低表达的MCF-7肿瘤模型来说,对半乳糖凝集素高表达的HepG2肿瘤明显更加偏爱,且随着时间的延长,在肝、肾中的比例逐渐下降,但在肿瘤组织中的比例逐渐增加。
经体内分布评价后,对化合物9、依托泊苷、奥沙利铂和5-氨基吲唑-鬼臼毒素进行了体内药效实验。
由图4、5、6、7可知,5-氨基吲唑-鬼臼毒素(5mg/kg)与依托泊苷(20mg/kg)展现出相近的杀灭肿瘤的效果,肿瘤较不给药组(Vehicle)明显缩小,肿瘤阻止塌陷,肿瘤细胞增殖蛋白表达降低等,化合物9进一步增大了这种差距。
经裸鼠体内药效评价后,对化合物9、依托泊苷、奥沙利铂和5-氨基吲唑-鬼臼毒素进行了小鼠体内安全性评价。
由图8、9、10、11、12可知,寡核苷酸的偶联明显改善了5-氨基吲唑-鬼臼毒素的毒性,且相比于依托泊苷、奥沙利铂,都表现出对器官的低毒性以及对血象降低的延缓等。
经裸鼠体内安全性评价后,对化合物9、依托泊苷和5-氨基吲唑-鬼臼毒素进行了C57小鼠体内药效评价。
由图13可知,化合物9的实验组表现出了明显的缓解肿瘤微环境中T细胞耗竭的能力,化合物9包括DBCO(二苯并环辛炔)修饰的寡核苷酸和5-氨基吲唑-鬼臼毒素,即二者发挥协同抗肿瘤效果,明显好于单独的5-氨基吲唑鬼臼毒素组和依托泊苷。
本实施例合成中间体3的反应单体5-硝基吲唑还可以替换为6-硝基-吲唑、7-硝基-吲唑、5-硝基-吲哚、6-硝基-吲哚、7-硝基-吲哚中的一种,因此合成的化合物3也对应变为
最终合成的化合物9为
其中,R
*为这些结构与氮原子的附接点;
R
R3为SEQ ID No.1所示的寡核苷酸序列,能够靶向肿瘤微环境中的半乳糖凝集素1。
- 烷基醇修饰的多西他赛衍生物抗癌药物化合物及其制备方法和应用
- 一类苯硼酸或其衍生物改性的多糖及其制备方法和应用
- 噻唑类衍生物及其制备方法和应用
- 一种2-巯基-5-氰基嘧啶类衍生物及其制备方法和应用
- 亲水性基团取代的吲哚甲酰胺类衍生物、其制备方法及其在医药上的应用
- 4β-氨基取代鬼臼毒素类衍生物及其制备方法和应用
- 4-硫取代鬼臼毒素类衍生物及其制备方法和应用