固体颗粒泡沫模板协同化学发泡制备的开孔吸附剂及其在快速去除废水中铀元素的应用
文献发布时间:2024-04-18 19:58:53
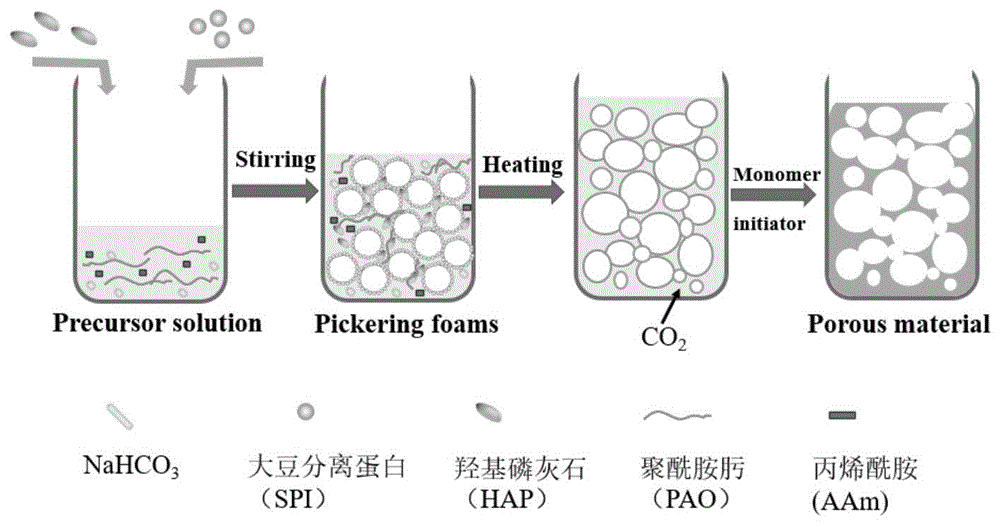
技术领域
本发明属于吸附剂领域,特别涉及固体颗粒泡沫模板协同化学发泡制备的开孔吸附剂及其在快速去除废水中铀元素的应用。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
核能的利用有效的缓解了能源短缺问题,但核工业的快速发展产生了大量放射性废水,铀元素作为废水中主要的放射性元素之一,在水性介质中易迁移,存在放射性高,毒性高的风险,因此放射性铀元素的去除与回收对保护生态环境、人体生命健康以及促进能源的可持续发展均具有重要意义。铀元素在不同的pH环境中呈现出不同的价态和存在方式,这也对其回收提出了不小的挑战。目前,常用的铀元素回收方法有离子交换法、超滤法、电化学法、絮凝法、吸附法等。其中,吸附法被认为是一种更为环保安全、操作方便、二次污染低、效率更高的方法。通常能够发挥吸附作用的功能性基团有氨基、磷酸基,酰胺肟等,其中,酰胺肟(AO)结构中的肟氮、氨基氮和肟氧都可以作为电子供体与铀酰离子进行配位,因此在多种离子共存的复杂废水环境中,酰胺肟被认为是提取铀酰离子(UO
发明内容
基于目前偕胺肟功能化的亲水大孔吸附剂传质效率低以及报道的互通孔吸附剂构筑方法能耗高,步骤繁琐,二次污染大的问题,本发明将泡沫模板和化学发泡法相结合,以一种更为节能环保的技术制备出了具有高渗透通量的偕胺肟功能化的开孔亲水吸附剂。
为了实现上述目的,本发明采用如下技术方案:
本发明的第一个方面,提供了一种固体颗粒泡沫模板协同化学发泡制备开孔吸附剂的方法,包括:
将大豆分离蛋白与羟基磷灰石加入到含有化学发泡剂、聚合单体、交联剂、热引发剂、聚偕胺肟的溶液中,涡旋混匀,搅拌发泡,得到固体颗粒稳定的泡沫样品;
将所述固体颗粒稳定的泡沫样品80℃~90℃水浴中加热25~35min,引发单体聚合和化学发泡剂热分解,反应完成后,烘干,得到开孔吸附剂;
其中,大豆分离蛋白、羟基磷灰石、化学发泡剂、聚合单体、交联剂、热引发剂、聚偕胺肟的质量比为0.1~0.2:0.2~0.4:0.1~0.2:0.18~0.36:0.75~1.5:0.020~0.040:0.75~1.5。
本发明采用了泡沫模板和化学发泡相结合的方法,泡沫作为一种气液分散体系,液体作为连续相,气体为分散相。该方法能够在液相引入聚合单体形成多孔材料的骨架,孔结构主要取决于泡沫气泡的大小与形状。无需除去分散介质或者制孔剂等后续步骤,是一种更为节能环保的方法。但以泡沫体系为模板制备材料的研究相对较少,主要原因是泡沫作为一种热力学不稳定的气液分散体系,在材料骨架形成过程中泡沫的稳定性很难维持,也就无法发挥泡沫模板的作用。与传统表面活性剂稳定的泡沫相比,由固体颗粒稳定的泡沫,颗粒通过有效的“不可逆吸附”在气液界面,可以提供更稳定的弹性屏障进而有效地防止气泡收缩和气体扩散。选择合适的颗粒稳定剂能够使得固体颗粒稳定的泡沫具有优异的稳定性,使其有可能用于制备泡沫材料。化学试剂的二次发泡有利于增加材料的孔隙率,暴露更多活性吸附位点并提升与目标物质的接触速度。
在一些实施例中,所述化学发泡剂为碳酸氢钠。
在一些实施例中,所述聚合单体为丙烯酰胺。
在一些实施例中,所述交联剂为N,N′-亚甲基双丙烯酰胺。
在一些实施例中,所述热引发剂为偶氮二异丁脒盐酸盐。
在一些实施例中,所述搅拌发泡的具体条件为18000r/min~22000r/min,发泡时间为0.8~1.2min。
在一些实施例中,所述聚偕胺肟的合成方法包括:将NH
更具体地,将大豆分离蛋白(SPI)与羟基磷灰石(HAP)加入到含有碳酸氢钠、丙烯酰胺(AAm)、交联剂N,N′-亚甲基双丙烯酰胺(MBA)与聚偕胺肟(PAO)的溶液中,在高速搅拌下利用双亲性颗粒大豆分离蛋白稳定气液界面得到固体颗粒稳定的泡沫,然后将该泡沫样品在80℃下加热,在单体聚合形成材料骨架的同时使得液相中的碳酸氢钠热分解产生二氧化碳进行二次发泡,那么,在单体聚合得到固体骨架的过程中颗粒稳定的泡沫与二氧化碳产的气泡均可达到造孔的目的,从而得到开孔以及连通孔结构的材料。羟基磷灰石也作为材料骨架的一部分,提升材料机械强度的同时与PAO共同发挥吸附功能。从而改善颗粒类材料循环使用困难以及酰胺肟块体材料理论吸附容量低的问题。
本发明的第二个方面,提供了上述的方法制备的开孔吸附剂。
本发明的第三个方面,提供了上述的开孔吸附剂在快速去除废水中铀元素中的应用。
在一些实施例中,所述废水的pH值为2-8。
本发明的有益效果
(1)本发明从构效关系出发,创新性的利用环保简便的固体颗粒稳定的泡沫模板协同化学发泡法制备出了具有互通的开孔结构吸附剂,改善了目前亲水性材料结构构筑方面的问题。亲水性开孔结构的引入,使得材料能够充分暴露吸附位点,HAP、PAO能够与铀离子进行充分的接触,提升了材料的吸附速率与吸附容量。
(2)本发明使用的两亲性颗粒SPI成本低廉,合成简单,HAP作为一种亲水的功能性矿物颗粒,能够发挥吸附功能的同时加固材料的骨架,与亲水性单体一起赋予材料亲水性,进而加速吸附活性位点与酰铀离子的结合进程。
(3)本发明制备方法简单、实用性强,易于推广。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示例性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
图1为本发明中亲水性开孔结构吸附剂制备过程示意图;
图2为实施例1中两亲性颗粒SPI及功能性吸附颗粒HAP的形态表征结果;其中,A为两亲性颗粒SPI的TEM图;B为两亲性颗粒SPI的粒径分布图;C为两亲性颗粒SPI的等电点电位图;D为功能性吸附颗粒HAP的TEM图;E为功能性吸附颗粒HAP的粒径分布图;F为功能性吸附颗粒HAP的等电点电位图;G为两亲性颗粒SPI及功能性吸附颗粒HAP的表面活性结果;H为两亲性颗粒SPI及功能性吸附颗粒HAP的固液气三相接触角表征结果;
图3为功能性聚合物的表征结果;A为PAN-PAO反应过程示意图;B为所述功能性聚合物的傅里叶红外变换谱图;
图4为两亲性颗粒SPI参与构筑的固体颗粒稳定的泡沫样品性质;其中,A为实物图,a、a
图5为功能性颗粒HAP与功能性聚合物PAO制备出了泡沫的流变学表征;
图6为泡沫样品稳定性考察,其中,A为实物图,a
图7为样品在80℃下放置30min的实物图及样品的光学显微镜照片。a、b、c为发泡后的样品,组成分别为a:SPI-0.15g/g,b:SPI-0.05g/g,HAP-0.1g/g,c:SPI-0.05g/g,HAP-0.1g/g,PAO-3wt%;
图8为SPI/HAP/PAO/NaHCO
图9为化学发泡法、颗粒稳定的泡沫模板法、化学发泡协同颗粒稳定的泡沫模板法得到的样品的SEM图(样品组成:a为HAP/PAO/NaHCO
图10为泡沫模板、化学发泡协同泡沫模板得到的样品进行压汞实验结果;
图11中a不同pH下SPI/HAP/PAO/NaHCO
图12为化学发泡协同颗粒稳定的泡沫模板法构筑的开孔吸附剂的动力学;
图13为化学发泡协同颗粒稳定的泡沫模板法构筑的开孔吸附剂吸附等温线并进行模型拟合;
图14为化学发泡协同颗粒稳定的泡沫模板法构筑的开孔吸附剂的选择吸附能力以及循环使用情况;
图15为吸附前后样品的XPS(a),XRD(b),FTIR(c)谱图以及N/O/Ca/P元素的高分辨XPS谱图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
实施例1聚偕胺肟的合成
将4.17g NH
实施例2两亲性颗粒SPI稳定的泡沫
取一定量的N,N′-亚甲基双丙烯酰胺(MBA)在80℃下溶解于水中得到1%wt的MBA水溶液备用。取1.5g质量分数为1%的MBA溶液于7mL塑料离心管中,将0.18g聚合单体AAm,0.020g热引发剂偶氮二异丁脒盐酸盐(AIBA)溶解于MBA溶液中,将0.3g大豆分离蛋白颗粒分散于上述水溶液中,涡旋30s使其分散均匀。将上述样品用高速分散器进行发泡,设置转速恒定为20000r/min,发泡1min即可得到由两亲性颗粒稳定的固体颗粒稳定的泡沫样品。不同浓度两亲性颗粒SPI稳定的泡沫制备方法与上述步骤相同。
实施例3含有HAP与PAO的固体颗粒稳定的泡沫
含有具备吸附能力的HAP以及PAO泡沫的制备:将PAO加入到0.15mol/L的氢氧化钠溶液中并在80℃下加热30min溶解,获得质量分数为6%的PAO前体溶液备用。取溶解好的PAO溶液0.75g与0.75g MBA水溶液混合均匀于7mL塑料离心管中,将0.18g聚合单体AAm,0.020g热引发剂偶氮二异丁脒盐酸盐(AIBA)加入上述混合溶液中,将0.1g大豆分离蛋白与0.2g羟基磷灰石颗粒分散于上述水溶液中,涡旋30s使其分散均匀。将上述样品用高速分散器进行发泡,设置转速恒定为20000r/min,发泡1min即可得到由两亲性颗粒稳定的含有HAP与PAO的固体颗粒稳定的泡沫样品。
实施例4含有HAP与PAO和化学发泡剂的固体颗粒稳定的泡沫
含有化学发泡剂碳酸氢钠的泡沫样品制备:将0.75g PAO溶液与0.75gMBA水溶液与于7mL塑料离心管中混合均匀后加入0.1g碳酸氢钠溶解,将0.18g聚合单体AAm,0.020g热引发剂偶氮二异丁脒盐酸盐(AIBA)加入上述混合溶液中,将0.1g大豆分离蛋白与0.2g羟基磷灰石颗粒分散于上述水溶液中,涡旋30s使其分散均匀。将上述样品用高速分散器进行发泡,设置转速恒定为20000r/min,发泡1min即可得到由两亲性颗粒稳定的含有HAP与PAO和化学发泡剂的固体颗粒稳定的泡沫样品。
实施例5吸附剂样品的制备
将实施例2-4中得到的一系列泡沫样品放入到80℃水浴锅中加热30min,在热引发AAm单体聚合的同时诱发碳酸氢钠热分解产生二氧化碳,在单体聚合形成骨架的同时达到二次制孔的目的。将聚合后的样品于烘箱烘干掉多余的水分,得到吸附剂样品,进行后续的表征和吸附实验。
实验例1颗粒的表征
对使用的两种颗粒的微观形态,等电点,表面活性,固液气三相接触角进行了表征,相关结果见图2。
通过图2以及粒径统计结果看出,两亲性颗粒SPI是尺寸在10nm左右的不规则球形结构,功能性吸附颗粒HAP是80nm左右的棒状结构,两者的等电点均是酸性的。通过表面张力测试发现,SPI具有降低表面张力的能力,HAP几乎没有表面活性。通过三相接触角测试发现,SPI与液-气的接触角在90°附近,说明其对水-气两相具有近似的润湿性,有稳定泡沫的潜力,并且SPI能够调节亲水性颗粒HAP与气液的接触角。
实验例2功能性聚合物的表征
相关结果见PAN-PAO反应过程及傅里叶红外变换谱图(FTIR)(图3)。
由图3可以看出,反应后的物质在2242cm
实验例3固体颗粒稳定的泡沫的制备
首先对只由两亲性颗粒参与构筑的固体颗粒稳定的泡沫样品性质进行了探究。相关结果见图4,分别为SPI浓度为0.03g/g,0.11g/g,0.17g/g稳定的固体颗粒稳定的泡沫的样品实物图,显微镜照片与流变学表征。
可以看出,在这三个浓度下均能形成由颗粒稳定的泡沫,并且通过应力-应变扫描,本发明可以看出随着颗粒用量增大泡沫的屈服应力点增大,对抗外力的能力增强。由频率扫描可以看出,三个样品的储能模量(G’)均大于损耗模量(G”),呈现出类固体状态。通过稳态剪切可以分析样品的黏度,可以看出,两亲性颗粒用量增加后,样品粘度增大,因而可以减缓排液速度,增加泡沫的稳定性。
本发明固定了两亲性颗粒的用量,向样品中加入功能性颗粒HAP与功能性聚合物PAO制备出了泡沫,并对样品进行了流变学表征,相关结果见图5(SPI,SPI/HAP,SPI/HAP/PAO泡沫样品的应力-应变扫描,频率扫描,稳态剪切)通过分析本发明发现,当两亲性颗粒用量固定时,额外加入的功能性矿物颗粒与聚合物均能增强样品的屈服应力数值与黏度,增加了泡沫样品的稳定性。
由于泡沫的稳定性对应用泡沫模板构筑材料的影响很大,本发明对后续将要进行大孔材料制备的泡沫样品稳定性进行了考察,相关结果见图6:样品实物图(a,b,c分别为未发泡、发泡后样品以及室温放置4h后的泡沫样品,样品组成分别为a
实验例4吸附剂的制备与表征
对前面得到的稳定的泡沫进行加热引发液相的单体聚合,烘干掉多余的水份得到固体材料,图8为样品的实物图以及样品的吸水情况,在1min内即可吸附高达自身重量20倍的水,具有快速吸水的能力,有利于材料在水溶液对目标物质进行一个快速的吸附。为了探究双发泡方法对孔结构的影响,本发明利用SEM对不同发泡方式得到的样品孔结构进行了观察,见图9:分别化学发泡法、颗粒稳定的泡沫模板法、化学发泡协同颗粒稳定的泡沫模板法得到的样品(样品组成分别为HAP/PAO/NaHCO
实验例5吸附实验的开展
由于酰铀离子的存在形态对环境pH的依赖很大,因此首先探究了pH对材料吸附能力的影响,在5mL含85ppm铀元素溶液中放置0.01g吸附剂,200r/min(所有吸附实验均固定在此转速)搅拌8h,通过测试吸附后铀元素浓度发现在酸性条件下吸附能力更强,对材料的Zeta电位进行测定发现,材料在一个宽的pH范围内均呈现负电性,因而本发明推测材料在酸性条件下吸附能力更优的原因是:酸性条件下铀离子是带正电的,与带负电的吸附剂之间存在静电吸引,从而利于酸性条件下对其进行吸附。本发明对不同材料组成和发泡手段得到的吸附剂的吸附能力进行了对比(50mL 280ppm溶液,0.02g吸附剂,吸附8h),得出SPI具备一定的吸附能力,HAP和PAO的加入均对材料整体的吸附能力进行了增强,并且加入碳酸氢钠发泡后,由于开孔结构的存在,吸附位点暴露程度增大,吸附能力得到了进一步的提升。相关结果见图11:不同pH下材料对酰铀离子的吸附情况,不同pH下材料的Zeta电位,不同组成与发泡手段构筑的材料对酰铀离子吸附情况的对比。
表1
本发明以化学发泡协同颗粒稳定的泡沫模板法构筑的开孔吸附剂为模型,对该吸附进程的吸附动力学(100mL 85ppm溶液,0.035g吸附剂吸附8h)、吸附等温线(50mL不同初始浓度的含铀溶液,0.02g吸附剂吸附4h,初始浓度分别为56,112.8,225.6,282,470,570,800ppm)、吸附热力学(50mL 300ppm含铀溶液,0.02g吸附剂吸附4h)进行了考察,并进行了相关的模型拟合。如图12(吸附剂的动力学进程)及表1(动力学模型拟合)所示,材料在4h内即可达到吸附平衡,并且通过模型拟合可以看出,伪二级动力学模型的R
表2
实验例6吸附机理的考察
相关结果见图15:吸附前后样品的XPS,XRD,FTIR谱图以及N/O/Ca/P元素的高分辨XPS谱图的分峰拟合。
通过对XPS分峰拟合得到的谱图可以看出,N/O/Ca/P元素的均向高结合能位置移动,说明功能性聚合物PAO上的氨基氮、肟氮以及肟氧能够作为供电子基团与酰铀离子的中心铀元素进行结合,功能性颗粒HAP中的Ca
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种用于去除核素铀的磁性吸附剂的制备及应用方法
- 去除水中抗生素的颗粒状吸附剂的制备方法、制得吸附剂及应用
- 去除水中抗生素的颗粒状吸附剂的制备方法、制得吸附剂及应用