一种普拉洛芬的精制方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明涉及药物的生产领域,具体涉及一种普拉洛芬的精制方法。。
背景技术
普拉洛芬系三环类的丙酸衍生物,化学名称为2-5H-[1]苯并吡喃[2,3-b]吡啶-7-基丙酸,含有两个镜像异构体,市售产品为消旋体。是welfide公司(原吉福制药株式会社,现三菱制药)开发的非甾体消炎镇痛药物。
对于普拉洛芬的制备,当前主流工艺是以5H-[1]苯并吡喃[2,3-b]吡啶为原料,经2-氯丙酰氯取代得到7-(2-氯丙酰基)-5H-[1]苯并吡喃[2,3-b]吡啶,在碱性条件下同时完成缩酮和水解两个反应,随后在磺酰氯的存在下发生重排,再经过水解制得普拉洛芬。该工艺得到的普拉洛芬粗品纯度一般可达到92%-98%,含有较多的杂质。与此同时,普拉洛芬对光、氧气等因素都敏感,所以除了后续制剂方向需对该产品的稳定性进行系统研究,同时也需要原料中的杂质能尽可能的降低。
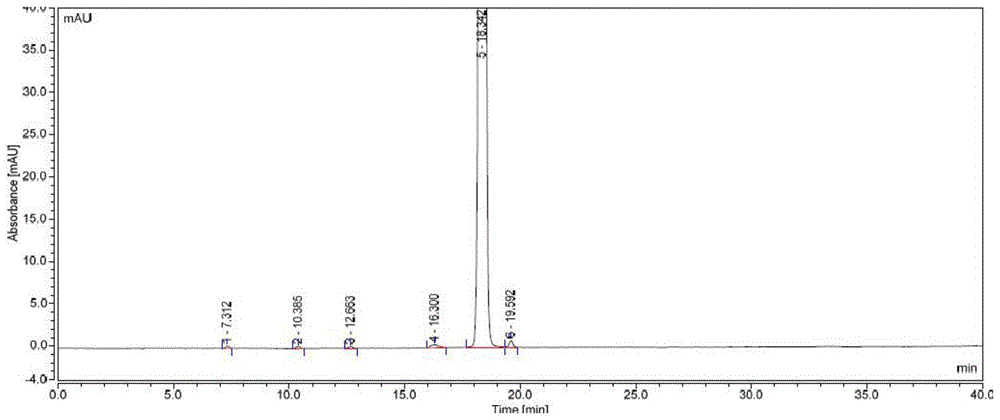
普拉洛芬中可能存在的杂质包括起始原料、中间体、降解产物、工艺杂质以及一些未知杂质等等。这些杂质以常规的重结晶精制方法可将大部分杂质控制在限度要求内,但有个别杂质较难控制。例如图1所示,保留时间紧邻普拉洛芬之后的杂质A,是氯代重排反应中消除副反应生成的烯烃杂质(式I);该杂质含量相对较高,很难通过工艺控制实现杂质的清除;同时,该杂质结构与普拉洛芬相似,普拉洛芬直接进行重结晶精制无法清除杂质A。又如图1所示,主峰前的杂质B,其保留时间为16.18min,该杂质为普拉洛芬的氧化杂质,在生产中会持续产生杂质B。
在已报道的方法中有提到乙醇-水重结晶、甲醇-水打浆然后甲醇-水重结晶、二氧六环-水重结晶等不同的精制方式,具体见下表;大多报道的精制方法没有较好的收率,仅个别精制收率为80%左右。经考察,文献中报道的方法均不能实现有效的精制,且收率普遍较低。如JPH03271290A、JPH0495094A报道的精制方法无法实现重结晶,打浆则无精制效果;US3931205A、CN110372713B报道的方法未见实质精制可能,且收率损失较大,两种精制方法的收率为40~55%。
/>
发明内容
针对现有技术的不足,本发明提供一种普拉洛芬的精制方法。
本发明的技术方案如下:一种普拉洛芬的精制方法,包括以下步骤:
a)普拉洛芬粗品在溶剂A中加热回流至溶清;
b)加入碱A或盐A的溶液,继续加热搅拌,使普拉洛芬成盐,冷却析晶;
c)过滤获得普拉洛芬盐,用溶剂A淋洗滤饼,抽干;
d)滤饼在溶剂B搅拌至溶清,过滤得滤液,滤液用酸调节pH,析出固体;
e)过滤、干燥得高纯度普拉洛芬;
在一些实施方案中,所述步骤a)中溶剂A选自乙醇、丙酮、甲醇中的一种或多种,优选为乙醇。
在一些实施方案中,所述步骤a)中溶剂A与普拉洛芬粗品的体积质量比为6:1。
在一些实施方案中,所述步骤b)中碱A选自氢氧化钠或氢氧化钾,优选为氢氧化钠;所述盐A选自氯化镁。
在一些实施方案中,所述普拉洛芬粗品与碱A或盐A的摩尔比为1:1~1.1。
在一些实施方案中,所述碱A或盐A的溶液与普拉洛芬粗品的体积质量比为2:1。
在一些实施方案中,所述步骤b)中析晶条件为冷却至20℃,析晶2小时。
在一些实施方案中,所述步骤c)中所用溶剂A的体积与普拉洛芬粗品的质量比为1:1。在一些实施方案中,所述步骤d)中所用溶剂B的体积与普拉洛芬粗品的质量比为8:1
在一些实施方案中,所述步骤d)中酸选自盐酸或乙酸,调节pH至4~5。
在一些实施方案中,所述步骤e)中干燥温度为50℃。
本发明的有益效果:本发明提供一种普拉洛芬的精制纯化方法,通过成盐增加普拉洛芬和杂质间的性质差异,可以有效去除杂质A和杂质B,制备的普拉洛芬纯度高,该纯化方法操作简便,且收率较高。
1.本发明普拉洛芬的精制纯化方法是一种简单易行、低成本的精制纯化方法,可有效降低普拉洛芬中的杂质,满足制剂需求。
2.本发明方法得到的普拉洛芬成品,其色谱纯度能够高于99.90%,其中杂质A含量小于0.10%,杂质B含量小于0.10%,总杂质小于0.10%,单次精制收率>90%。
3.本发明中成盐步骤对各杂质的去除均有显著的效果,尤其是对杂质A的清除方面,效果十分显著,一次成盐可降低70~80%左右的杂质A。
附图说明
图1是实施例未处理过的普拉洛芬粗品的杂质色谱图
图2是本发明实施例1的普拉洛芬的杂质色谱图
图3是本发明实施例2的普拉洛芬的杂质色谱图
图4是本发明实施例3的普拉洛芬的杂质色谱图
图5是本发明对比例1的普拉洛芬的杂质色谱图
图6是本发明对比例2的普拉洛芬的杂质色谱图
图7是本发明对比例3的普拉洛芬的杂质色谱图
图8是本发明对比例4的普拉洛芬的杂质色谱图
具体实施方式
下面的实施例可以使本专业的本领域技术人员更全面地理解本发明,但并不因此将本发明限制在所述的实施例范围之中。
在实施例和对比例中使用的检测方法如下表所示,该方法参照高效液相色谱法(中国药典2020年版通则0512)测定。避光操作。
在实施例中,所用的需要精制纯化的普拉洛芬粗品参考“《普拉洛芬的合成研究改进》(精细化工中间体2009,39(3),3739)”制备,其制备方法为:以5H-[1]苯并吡喃[2,3-b]吡啶为原料,二氯甲烷(10V)为反应溶剂,在三氯化铝(2.5eq)存在的条件下,同2-氯丙酰氯(1.2eq)发生傅克酰基化反应得到7-(2-氯丙酰基)-5H-[1]苯并吡喃[2,3-b]吡啶;随后在甲醇(5V)-甲醇钠(1.5eq)的条件下制备缩酮并水解得到1-(5H-苯并吡喃[2,3-b]吡啶-7-基)-1,1-二甲氧基丙基-2-醇;1-(5H-苯并吡喃[2,3-b]吡啶-7-基)-1,1-二甲氧基丙基-2-醇再在磺酰氯(1.2eq)、三乙胺(2.4eq)的存在下发生重排获得普拉洛芬甲酯,最后经过水解、后处理制得普拉洛芬粗品。
未精制的普拉洛芬粗品的HPLC谱图见图1所示,色谱纯度为98.78%,其中主要杂质含量为:杂质A 0.23%,杂质B 0.11%,总杂质1.22%。
具体测试数据如下表所示:
实施例1
将普拉洛芬粗品(40.00g,0.157mol)加6倍量的乙醇(240mL),升温至80℃,体系回流。加入溶有氢氧化钠(6.14g,0.154mol)的2倍量乙醇(80mL)溶液,继续加热搅拌;回流3-5分钟,固体析出,冷却至20℃,析晶2小时。过滤,用乙醇(40mL)淋洗滤饼,抽干。固体返回反应瓶中,加入的8倍量的水(320mL),搅拌至溶清,过滤得滤液,滤液用乙酸调pH至4~5,固体析出,过滤,抽干得湿品,于50℃烘干,得普拉洛芬成品39.95g,收率92.0%。产品的色谱图如图2所示,其色谱纯度为99.91%,其中杂质A 0.03%,杂质B0.02%,总杂0.09%。具体实验数据如下表所示:
实施例2
将普拉洛芬粗品(40.00g,0.157mol)加8倍量的丙酮(320mL),升温至60℃,体系回流,加入溶有氢氧化钾(8.61g,0.154mol)的2倍量乙醇(80mL)溶液,继续加热搅拌,3~5分钟后固体析出,冷却至20℃,析晶2小时。过滤,并用丙酮淋洗滤饼,抽干;滤饼返回反应瓶中,加入8倍量的水(320mL),搅拌至溶清,过滤得滤液,滤液用乙酸调pH至4~5,固体析出,过滤,抽干得湿品,湿品于50℃烘干,得普拉洛芬成品39.13g,收率90.1%。产品的色谱图如图3所示,其色谱纯度为99.84%,其中杂质A 0.05%,杂质B 0.05%,总杂0.16%。具体实验数据如下表所示:
实施例3
将普拉洛芬粗品(20.00g,0.078mol)加6倍量的甲醇(120mL),升温至65℃,体系回流,加入溶有氯化镁(7.31g,0.072mol)的2倍量甲醇(40mL)溶液,继续加热搅拌,回流3~5分钟,固体析出,冷却至20℃,析晶2小时。过滤,并用甲醇淋洗滤饼,抽干;滤饼返回反应瓶中,加入8倍量的水(160mL),搅拌至溶清,过滤得滤液,滤液用盐酸调pH至4~5,固体析出,过滤,抽干得湿品,湿品于50℃烘干,即得普拉洛芬成品19.94g,收率91.9%。产品的色谱图如图4所示,其色谱纯度为99.78%,其中杂质A 0.071%,杂质B 0.04%,总杂0.22%。具体实验数据如下表所示:
与此同时,参考各类文献对实施例中同一批次普拉洛芬粗品进行了精制,具体结果见对比例1~对比例4。
对比例1
参考文献:金荣庆,张海波,孟霆,等.普拉洛芬的合成研究改进[J].精细化工中间体,2009,39(3):4.
将普拉洛芬粗品(10.00g,0.039mol),加5倍量乙醇(50mL),氮气保护下加热搅拌至回流,体系溶清;氮气保护下加入纯化水(50mL),保温搅拌30min,缓慢冷却至20℃,析晶2小时。过滤,并用少量乙醇淋洗滤饼,抽干,滤饼于50℃烘干,即得普拉洛芬成品5.12g,收率51.2%。产品的色谱图如图5所示,其色谱纯度为98.80%,其中杂质A 0.16%,杂质B0.015%,总杂1.20%。具体实验数据如下表所示:
对比例2
参考文献:饶汉才,黄能章,王硕,等.一种普拉洛芬的精制纯化方法.CN110372713B.
将普拉洛芬粗品(15.00g)研细过80目筛,取(10.00g,0.039mol)加8倍量(80mL)的50%的甲醇-水溶液,升温至60-65℃回流并保温搅拌4h,冷却至室温,过滤,以少量50%的甲醇-水溶液淋洗滤饼,抽干,低温(30℃)烘干,得产物6.13g,打浆收率61.3%。取打浆处理好的普拉洛芬,加25倍量的50%的甲醇-水溶液置三口瓶中,加热至回流,搅拌使溶解,回流30分钟进行脱色,趁热过滤,滤液加入干净的三口瓶中,继续搅拌30分钟,冷却至25℃,析晶5h。过滤,用少量重结晶溶剂(50%甲醇溶液)淋洗滤饼,抽干,于55℃烘干,得普拉洛芬成品4.25g,两次操作总收率42.5%。产品的色谱图如图6所示,其色谱纯度为99.26%,其中杂质A 0.21%,杂质B未检出,总杂0.74%。具体实验数据如下表所示:
由以上样品有关物质的HPLC谱图数据同普拉洛芬粗品的HPLC谱图数据比对可知,CN110372713B提供的精制方法对普拉洛芬粗品存在一定的精制效果,但对18.5min的杂质A没有精制效果,对4.8min和6.0min的杂质无法实现有效去除,整个操作过程的收率较低。
对比例3
将普拉洛粗品(10.00g,0.039mol),加3倍量二甲亚砜(30mL),室温搅拌至体系溶清,滴加水至体系恰好析出固体,冷却至0~10℃,析晶1~2小时。过滤,并用少量水淋洗滤饼,抽干,滤饼于50℃烘干,即得普拉洛芬成品5.17g。产品的色谱图如图7所示,其色谱纯度为98.99%,其中杂质A 0.17%,杂质B 0.005%,总杂0.49%,收率51.7%。具体实验数据如下表所示:
对比例4
参考文献:MITSUBISHI CHEMICAL HOLDINGS CORPORATION.Substitutedalkanoic acids and derivatives.US3931205,1976,A
将普拉洛粗品(10.00g,0.039mol),加5倍量二氧六环(50mL),室温搅拌至体系溶清,滴加水至体系恰好析出固体,冷却至0~10℃,析晶1~2小时。过滤,并用少量水淋洗滤饼,抽干,滤饼于50℃烘干,得普拉洛芬7.23g。将烘干的物料再次返回反应瓶中,加5倍量二氧六环(36mL),室温搅拌至体系溶清,滴加水至体系恰好析出固体,冷却至0~10℃,析晶1~2小时。过滤,并用少量水淋洗滤饼,抽干,滤饼于50℃烘干,即得普拉洛芬成品5.13g,收率51.3%。产品的色谱图如图8所示,其色谱纯度为99.70%,其中杂质A 0.18%,杂质B ND,总杂0.30%。具体实验数据如下表所示:
由上述对比例可知,一般普拉洛芬的重结晶工艺可以去除其他部分杂质;但对于杂质A普通的重结晶工艺对该杂质的去除作用非常有限,即使重复精制操作也不能有效降低杂质A的含量,而收率会大幅度降低。而本发明中的精制工艺则对各杂质有显著的去处效果,尤其是对杂质A的清除方面,获得了意想不到的效果,可确保产品符合预期的质量要求。