一种利非斯特中间体苯并呋喃-6-羧酸的制备方法
文献发布时间:2024-04-18 19:59:31
技术领域
本发明涉及药物技术领域,尤其涉及一种利非斯特中间体苯并呋喃-6-羧酸的制备方法。
背景技术
干眼症是由任何原因引起的泪液质和量异常或动力学异常导致的泪膜稳定性下降,并伴有眼部不适,导致眼表组织病变为特征的多种疾病的总称,为常见的眼科疾病,轻度干眼症将会对我们的工作以及生活造成影响,重度干眼症则会导致失明。干眼症的发病机制复杂且尚未完全明确,干眼症的分类主要分为水液缺乏型、脂质异常型、黏蛋白异常型、泪液动力学异常型以及由炎症引发的干眼症等。由炎症导致的干眼症主要由于中性粒细胞介导,通过激活丝裂原活化蛋白激酶和核因子活化B细胞K-轻链增强通路,从而出现炎性细胞因子,从而导致炎症的产生。
利非斯特(lifitegrast)是一种治疗所述的干眼症的重要的药物,是新型的小分子整合素(integrin)抑制药,能拮抗淋巴细胞功能相关抗原-1(LFA-1),阻断与其同源配体细胞间黏附分子-1(ICAM-1)的相互作用,干扰引起干眼病的角膜与结膜组织的ICAM-1过度表达(陈本川.治疗干眼症新药——利非斯特(lifitegrast)[J].医药导报,2017,36(2):6)。苯并呋喃-6-羧酸是制备利非斯特关键中间体。
现有文献公开了一些利非斯特或苯并呋喃-6-羧酸合成方法:
Organic Syntheses(DOI:10.15227/orgsyn.046.0028)公布了苯并呋喃的一种制备方法。以水杨醛为原料,经氯乙酸取代,关环得到苯并呋喃。具体合成路线如下:
该方法制备苯并呋喃的方法路线较短,但合成的收率较低,第二步的最高收率仅为67.8%。
美国专利US16247843公开了一种LFA-1抑制剂的制备方法,其公开的苯并呋喃-6-羧酸制备方法为以6-羟基-2H-苯并呋喃-3-酮为原料,经叔丁基二甲基氯硅烷保护羟基、还原、脱保护、引入羧基等反应得到苯并呋喃-6-羧酸,具体合成路线如下:
此方法的缺点在于:第一,路线较为复杂;第二,需要用到醋酸钯,存在重金属残留超标的风险;第三,引入羧基时需要使用高压,且需要通一氧化碳气体,不利于安全生产;第四,反应时间长。
中国专利CN110684000公开了一种制备方法,以3-羟基苯甲酸为原料,经六亚甲基四胺引入醛基、酯化、witting反应关环、水解得到苯并呋喃-6-羧酸。具体合成路线如下:
该方法的缺点在于:第一,反应路线较长,总收率仅为19.0%;第二,六亚甲基四胺是《易制爆危险化学品名录》的管制品、二溴甲烷为危险品,试剂购买不方便;第三,高温反应时间为24h以上,不利于工业生产。
可以看到,目前制备利非斯特中间体苯并呋喃-6-羧酸的方法普遍存在着不够经济的缺陷,制备过程复杂、初始原料昂贵、反应条件苛刻、副产物不易控制及除去,这些问题将会提高利非斯特的合成成本,本领域迫切需要成本更低的苯并呋喃-6-羧酸制备方法。
发明内容
为了解决上述问题,本发明提供了一种成本低、收率高、反应条件温和的利非斯特中间体苯并呋喃-6-羧酸的制备方法。
为了实现上述发明目的,本发明提供了一种利非斯特中间体苯并呋喃-6-羧酸的制备方法,包括以下步骤:
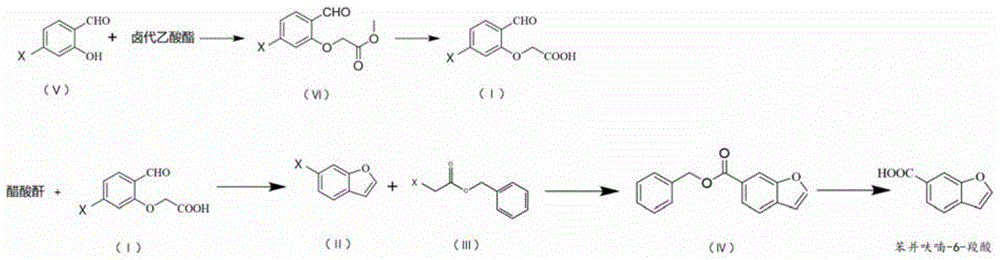
S1,式(Ⅰ)化合物与醋酸酐在加热条件下进行关环反应,得到式(Ⅱ)化合物;
其中,X为卤素;
S2,式(Ⅱ)化合物与式(Ⅲ)化合物在正丁基锂的催化下进行催化反应得到式(Ⅳ)化合物,再在碱性条件下进行第一水解反应,得到苯并呋喃-6-羧酸;
其中,X为卤素。
优选地,步骤S1中,加热的温度为100~150℃,关环反应的时间为5~12h。
优选地,步骤S1中,式(Ⅰ)化合物的质量与醋酸酐的体积之比为1:1~20。
优选地,步骤S2中,催化反应在保护气的保护下进行,催化反应的温度为-78~-20℃;式(Ⅱ)化合物与式(Ⅲ)化合物的摩尔比为1:1.5~2.5。
优选地,步骤S2中,碱性条件为pH≥12;
优选地,催化反应结束后,向反应液中加入碱性物质调节pH值,进行第一水解反应,得到苯并呋喃-6-羧酸。
优选地,式(Ⅰ)化合物、式(Ⅱ)化合物中的X为溴,式(Ⅰ)化合物中的X为氯。
优选地,式(Ⅰ)化合物的制备方法包括以下步骤:
其中,X为卤素;
A1,式(Ⅴ)化合物与卤代乙酸酯在加热条件下反应,得到式(Ⅵ)化合物;
A2,式(Ⅵ)化合物在碱性条件下进行第二水解,得到式(Ⅰ)化合物。
进一步优选地,式(Ⅰ)化合物的制备方法包括以下条件中的一种或多种:
(a)步骤A1中,加热的温度为40~100℃;
(b)步骤A1中,卤代乙酸酯选自氯乙酸甲酯、氯乙酸乙酯、氯乙酸苄酯和氯乙酸异丙酯中的一种或多种;
(c)步骤A1中,式(Ⅴ)化合物与卤代乙酸酯的摩尔比为1:1~2;
(d)步骤A1中,反应的时间为1~6h;
(e)步骤A2中,碱性条件为pH≥12;
(f)步骤A2中,将式(Ⅵ)化合物与碱性溶液混合进行第二水解。
进一步优选地,条件(f)所述的碱性溶液选自氢氧化钠溶液、碳酸钠溶液、碳酸钾溶液或碳酸铯溶液;
和/或,式(Ⅵ)化合物与碱性溶液的摩尔比为1:2~4。
进一步优选地,式(Ⅴ)化合物和式(Ⅵ)化合物中的X为溴。
本发明还提供了一种用于制备利非斯特或苯并呋喃-6-羧酸的中间体化合物,包括如式(Ⅰ)所示的化合物:
其中,X为卤素。
与现有技术相比,本发明的有益效果:
1、通过本发明所述方法合成苯并呋喃-6-羧酸并未见报道,具有一定的新颖性;
2、本发明所述的苯并呋喃-6-羧酸制备方法以醋酸酐替代了醋酸钠及醋酸的使用,原子经济性更高,优化了关环反应条件使其更适宜工业化生产,得到的苯并呋喃-6-羧酸纯度可以达到90%以上,反应路线短、原料廉价易得,使反应更经济;
3、本发明所述的苯并呋喃-6-羧酸制备方法中避免管制试剂(醋酸酐为二类易制毒,工业化生产能够购买获得)、重金属、一氧化碳的使用,反应过程温和避免高压操作、高温加热时间在8h左右,有效降低了反应成本;
4、本发明所述的苯并呋喃-6-羧酸制备方法优化了关环条件,更适宜工业化生产且收率有较大幅度的提升,本发明所述制备方法总收率在75%以上(以式(Ⅴ)化合物计),而现有技术如专利CN104797574的收率仅仅为48.11%。
附图说明
图1为本发明所述制备方法的合成路线示意图;
图2为实施例1中式(Ⅰ)化合物的质谱图;
图3为实施例5式(Ⅱ)化合物的质谱图;
图4为实施例9式(Ⅴ)化合物的质谱图;
图5为实施例1中式(Ⅰ)化合物的氢谱图;
图6为实施例5式(Ⅱ)化合物的氢谱图;
图7为实施例9式(Ⅴ)化合物的氢谱图。
具体实施方式
本发明提供了本发明提供了一种利非斯特中间体苯并呋喃-6-羧酸的制备方法,包括以下步骤:
S1,式(Ⅰ)化合物与醋酸酐在加热条件下进行关环反应,得到式(Ⅱ)化合物;
其中,X为卤素;
本发明选择式(Ⅰ)化合物与醋酸酐进行关环反应,并对反应条件进行了优化,去除高压等成本较高的反应条件,有效提高关环反应的原子经济性,降低反应成本。在本发明的一些具体实施方式中,所述关环反应时的加热温度优选为100~150℃,更优选为115~140℃,进一步优选为130℃;所述关环反应的反应时间优选为5~12h,更优选为6~8h。在本发明的一些具体实施方式中,式(Ⅰ)化合物的质量与醋酸酐的体积之比优选为1:1~20,更优选为1:5。在本发明中,式(Ⅰ)化合物和式(Ⅱ)化合物中的取代基X选自F、Cl或Br;在本发明的一些具体实施方式中,式(Ⅰ)化合物中的取代基X为Br。
在本发明的一些具体实施方式中,式(Ⅰ)化合物可以采用包括下述步骤的方法制备得到:
其中,X为卤素;
A1,式(Ⅴ)化合物与卤代乙酸酯在加热条件下反应,得到式(Ⅵ)化合物;
A2,式(Ⅵ)化合物在碱性条件下进行第二水解,得到式(Ⅰ)化合物。
在本发明的一些具体实施方式中,步骤A1中所述加热的温度优选为40~100℃,更优选为60~75℃,进一步优选为70℃;步骤A1所述反应的时间优选为1~6h,更优选为2~3h。在本发明的一些具体实施方式中,步骤A1中所述的卤代乙酸酯包括但不限于氯乙酸甲酯、氯乙酸乙酯、氯乙酸苄酯和氯乙酸异丙酯中的一种或多种,优选为氯乙酸甲酯或氯乙酸乙酯,进一步优选为氯乙酸甲酯。在本发明的一些具体实施方式中,步骤A1中式(Ⅴ)化合物与卤代乙酸酯的摩尔比优选为1:1~2,更优选为1:1.2。
在本发明中,步骤A2所述的碱性条件优选为pH≥12。在本发明的一些具体实施方式中,按照步骤A1得到式(Ⅵ)化合物后,将其分离并与碱性溶液混合,进行第二水解,得到式(Ⅰ)化合物;所述的碱性溶液包括但不限于氢氧化钠溶液、碳酸钠溶液、碳酸钾溶液或碳酸铯溶液;优选地,式(Ⅵ)化合物与碱性溶液的摩尔比为1:2~4。在本发明的一些具体实施方式中,式(Ⅴ)化合物和式(Ⅵ)化合物中的取代基X可以是F、Cl或Br,优选为Br。
S2,式(Ⅱ)化合物与式(Ⅲ)化合物在正丁基锂的催化下进行催化反应得到式(Ⅳ)化合物,再在碱性条件下进行第一水解反应,得到苯并呋喃-6-羧酸;
其中,X为卤素。
本发明选择正丁基锂作为催化剂。在本发明的一些具体实施方式中,催化反应在保护气的保护下进行,保护气体可以是氮气,本发明的步骤S2中以保护气体替代了较高压力的反应条件;所述催化反应的温度优选为-78~-20℃,更优选为-70~-65℃;所述催化反应的反应时间优选为0.5~2h,更优选为1~1.5h。在本发明的一些具体实施方式中,式(Ⅱ)化合物与式(Ⅲ)化合物的摩尔比优选为1:1.5~2.5,更优选为1:2。
在本发明中,所述的碱性条件优选为pH值≥12。在本发明中,催化反应得到式(Ⅳ)化合物后无需分离,直接向反应液中加入碱性物质调节溶液pH值为碱性即可进行第一水解反应,得到苯并呋喃-6-羧酸;所述的碱性物质包括但不限于氢氧化钠、氢氧化钾或氢氧化锂。在本发明的一些具体实施方式中,式(Ⅰ)化合物中的取代基X可以是F、Cl或Br,优选为Cl。
在本发明中,制备苯并呋喃-6-羧酸的各个步骤的反应时间均可采用实验室常规的监测手段(如TLC监测)具体确定,根据监测选择是否继续反应,并且结束反应后根据需要选择是否进一步纯化等,本领域常用的纯化等手段均可适用于本发明
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
实施例1 2-(4-溴-2-甲烷酰-苯氧基)(Ⅰ)的合成
室温下,向三口烧瓶中加入4-溴-2-羟基苯甲醛(2,10g,49.75mmol)、碳酸钾(13.75g,99.50mmol)、碘化钾(8.26g,49.75mmol)以及乙腈(100ml)搅拌,待4-溴-2-羟基苯甲醛完全溶解后,加入氯乙酸甲酯(6.48g,59.70mmol),升温至70℃反应,反应2h后,冷却至室温,调pH值至3左右,用乙酸乙酯(100ml×2)萃取,合并有机相,有机相用饱和食盐水(100ml×2)洗涤,旋干得到黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯14.16g。
将得到的黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯溶于50ml甲醇中,加入2M的氢氧化钠溶液(100ml),室温搅拌1h,调pH至2~3,用乙酸乙酯(200ml×2)萃取,合并有机相,有机相减压旋干,得到黄色固体2-(4-溴-2-甲烷酰-苯氧基)(12.63g),收率98.0%。
m p 147.3~151.4℃,HPLC纯度为90.15%。ESI-MS m/z 256.89[M+H]+。1H NMR(400MHz,CDCl3):δ10.36(s,1H),7.74(d,J=8.3Hz,1H),7.34(dd,J=8.2,1.2Hz,1H),7.12(d,J=1.6Hz,1H),4.83(s,2H).
实施例2 2-(4-溴-2-甲烷酰-苯氧基)乙酸(Ⅰ)的合成
室温下,向三口烧瓶中加入4-溴-2-羟基苯甲醛(2,5g,24.87mmol)、碳酸钾(6.88g,49.75mmol)、碘化钾(4.95g,29.85mmol)以及乙腈(50ml)搅拌,待4-溴-2-羟基苯甲醛完全溶解后,加入氯乙酸乙酯(5.40g,49.75mmol),升温至回流,反应1h后,冷却至室温,调pH值至3左右,用乙酸乙酯(50ml×2)萃取,合并有机相,有机相用饱和食盐水(50ml×2)洗涤,旋干得到黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯5.32g。
将得到的黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯溶于30ml甲醇中,加入2M的氢氧化钠溶液(50ml),室温搅拌1h,调pH值,用乙酸乙酯(200ml×2)萃取,合并有机相,有机相旋干,得到黄色固体2-(4-溴-2-甲烷酰-苯氧基)乙酸(4.43g),收率98.85%。
m p 147.3~151.4℃,HPLC纯度为91.24%,ESI-MS m/z 256.89[M+H]+。
实施例3 2-(4-溴-2-甲烷酰-苯氧基)乙酸(Ⅰ)的合成
室温下,向三口烧瓶中加入4-溴-2-羟基苯甲醛(2,20g,99.49mmol)、碳酸钾(20.63g,149.24mmol)、碘化钾(33.03g,198.99mmol)以及乙腈(200ml)搅拌,待4-溴-2-羟基苯甲醛完全溶解后,加入氯乙酸苄酯(16.97g,99.49mmol),升温至40℃反应,反应6h后,冷却至室温,调pH值至2,用乙酸乙酯(100ml×2)萃取,合并有机相,有机相用饱和食盐水(100ml×2)洗涤,旋干得到黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯20.34g。
将得到的黄色固体2-(5-溴-2-甲酰基苯氧基)乙酸乙酯溶于100ml甲醇中,加入2M的氢氧化钠溶液(200ml),室温搅拌1h,调pH值至2,用乙酸乙酯(200ml×2)萃取,合并有机相,有机相旋干,得到黄色固体2-(4-溴-2-甲烷酰-苯氧基)乙酸(16.85g),收率94.02%。
m p 147.3~151.4℃,HPLC纯度为95.56%,ESI-MS m/z 256.89[M+H]+。
实施例4 2-(4-溴-2-甲烷酰-苯氧基)乙酸(Ⅰ)的合成
室温下,向三口烧瓶中加入4-溴-2-羟基苯甲醛(2,20g,99.49mmol)、碳酸钾(20.63g,149.24mmol)、碘化钾(33.03g,198.99mmol)以及乙腈(100ml)搅拌,待4-溴-2-羟基苯甲醛完全溶解后,加入氯乙酸苄酯(22.04g,119.39mmol),升温至40℃反应,反应6h后,冷却至室温,调pH值至3,用乙酸乙酯(100ml×2)萃取,合并有机相,有机相用饱和食盐水(100ml×2)洗涤,旋干得到黄色固体28.56g。
将得到的固体溶于200ml甲醇中,加入2M的氢氧化钠溶液(100ml),室温搅拌1h,调pH值至3,用乙酸乙酯(100ml×2)萃取,合并有机相,有机相旋干,石油醚打浆,得到黄色固体3(25.27g),收率98.03%。m p 147.3~151.4℃,HPLC纯度为95..56%,ESI-MS m/z256.89[M+H]+。
实施例5 6-溴苯并呋喃(Ⅱ)的合成
室温下,向三口烧瓶中加入2-(4-溴-2-甲烷酰-苯氧基)乙酸(3,10.39g,40.11mmol)、乙酸酐(50ml),搅拌并升温至130℃,反应8h后,温度降至室温,加水,调pH至9左右,用乙酸乙酯(50ml×2),合并有机相,有机相旋干,得到深棕色油状物,经柱层析分离得到无色液体6-溴苯并呋喃(4,7.62g),收率96.43%。HPLC纯度为96.34%。
ESI-MS m/z 214.94[M+18]+。1H NMR(400MHz,CDCl3)δ7.71(s,1H),7.62(d,J=2.2Hz,1H),7.48(d,J=8.3Hz,1H),7.38(dd,J=8.3,1.6Hz,1H),6.77(dd,J=2.2,0.9Hz,1H).
实施例6 6-溴苯并呋喃(Ⅱ)的合成
室温下,向三口烧瓶中加入2-(4-溴-2-甲烷酰-苯氧基)乙酸(3,40g,154.41mmol)、乙酸酐(400ml),搅拌并升温至150℃,反应5h后,温度降至室温,加水,调pH至9左右,用乙酸乙酯(400ml×2),合并有机相,有机相旋干,得到深棕色油状物,经柱层析分离得到无色液体(4,29.09g),收率95.62%。
HPLC纯度为97.12%,ESI-MS m/z 214.94[M+18]+。
实施例7 6-溴苯并呋喃(Ⅱ)的合成
室温下,向三口烧瓶中加入2-(4-溴-2-甲烷酰-苯氧基)乙酸(3,5g,19.30mmol)、乙酸酐(5ml),搅拌并升温至100℃,反应12h后,温度降至室温,加水,调pH至9左右,用乙酸乙酯(30ml×2),合并有机相,有机相旋干,得到深棕色油状物,经柱层析分离得到无色液体6-溴苯并呋喃(4,3.66g),收率96.15%,HPLC纯度为96.81%。
ESI-MS m/z 214.94[M+18]+。
实施例8 6-溴苯并呋喃(Ⅱ)的合成
室温下,向三口烧瓶中加入2-(4-溴-2-甲烷酰-苯氧基)乙酸(3,30g,115.81mmol)、乙酸酐(600ml),搅拌并升温至140℃,反应6h后,温度降至室温,加水,调pH至9左右,用乙酸乙酯(400ml×2),合并有机相,有机相旋干,得到深棕色油状物,经柱层析分离得到无色液体6-溴苯并呋喃(4,21.68g),收率95.02%。HPLC纯度为97.34%,ESI-MS m/z214.94[M+18]+。
实施例9苯并呋喃-6-羧酸的合成
在N
m p 186.8~188.2℃,HPLC纯度为94.30%,ESI-MS m/z 161.03[M-H]+。1H NMR(600MHz,CDCl3)δ8.30(s,1H),8.04(dd,J=8.1,1.3Hz,1H),7.80(d,J=2.1Hz,1H),7.68(d,J=8.1Hz,1H),6.86(dd,J=2.1,0.9Hz,1H).
实施例10苯并呋喃-6-羧酸的合成
在N
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。