一种胺基喹唑啉酮衍生物的制备方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于有机合成技术领域,尤其涉及一种胺基喹唑啉酮衍生物的制备方法。
背景技术
喹唑啉酮及其衍生物是许多替尼类天然药物的核心结构,也是中药常山碱的基本结构单元,主要存在于常山、大青叶等中药中,具有重要的生物活性,对表皮生长因子受体(EGFR)或其酪氨酸激酶(EGFR-TK)、血管内皮生长因子受体(VEGFR)、神经生长因子受体(NGFR)及其他多个作用靶点有抑制活性,发挥抗癌、抗病毒等多种药理作用。
由于此类药物具有优异的药理活性,因此以喹唑啉酮母核为基础的衍生物研究成为热点,特别是对4-喹唑啉酮类衍生物的结构合成和修饰建立简单、高效的合成喹唑啉酮衍生物的方法,目前报道的喹唑啉酮类化合物的合成方法主要有以下几种:
以邻氨基苯甲酸(化合物A)在醋酸酐或三乙氧基乙醚条件下,先合成类似于靛红酸酐(化合物B)的中间体,该中间体再与胺在乙酸条件下合成甲基喹唑啉酮(化合物C),进而对甲基喹唑啉酮修饰得到目标产物(J MED CHEM,2016,59(10):5011-5021.);
以邻氨基苯甲酰胺衍生物(化合物D)在三氟甲磺酸铜及配体的条件下环合生成目标产物(ORG BIOMOL CHEM,2017,15(34):7140-7146.);
以邻氨基苯甲酰胺(化合物E)与醛(化合物F)反应生成喹唑啉酮(化合物G),喹唑啉酮再与溴化物(化合物H)发生N-烷基化反应得到目标产物(BIOORG MED CHEM LETT,2017,27(15):3529-3533.;EUR J MED CHEM,2021,212:112996.);
以邻氨基苯甲酰胺(化合物E)与胺(化合物I)反应生成喹唑啉酮(化合物K),喹唑啉酮再衍生化反应得到目标产物(ChemistrySelect,2017,2(17):4963-4968.);
以邻氨基苯甲酰胺(化合物E)与丙酮酸(化合物J)反应生化合物K,化合物K再在碱性条件下关环得到喹唑啉酮(Molecules,2023,28(10):4240.);
此外还有采用邻氨基苯甲酸胺(化合物E)与羧酸反应(化合物L)(EUR J MEDCHEM,2021,212:112996.,2023:101597.)、与醇(化合物M)(Organometallics,2021,40(6):725-734.)反应构建喹唑啉酮。
而此类反应虽然能简单高效合成喹唑酮类化合物,底物适用范围广,但是还存在邻氨基苯甲酸是第一类易制毒化学品、底物合成困难、合成需要贵金属催化等问题。
发明内容
本发明实施例的目的在于提供一种胺基喹唑啉酮衍生物的制备方法,旨在解决上述背景技术中提出的问题。
本发明实施例是这样实现的,一种胺基喹唑啉酮衍生物的制备方法,包括以下步骤:在铜盐及碱催化下,将氰氨、邻卤苯甲酰胺进行反应,得到胺基喹唑啉酮类衍生物,反应式如下:
优选地,所述氰氨为氰氨化钙(石灰氮)、氰氨化锂、氰胺化钠、氰胺化钾、单氰胺、双氰胺中的一种。
优选地,所述氰氨为氰氨化钙或单氰胺。
优选地,所述邻卤苯甲酰胺中的L基为氟、氯、溴、碘、氨基中的一种,X基为氧或硫。
优选地,所述L基为氯、溴或碘。
优选地,所述邻卤苯甲酰胺和胺基喹唑啉酮类衍生物中的R
优选地,所述氰氨与邻卤苯甲酰胺的摩尔比为1:0.9~3.0,铜盐与邻卤苯甲酰胺的摩尔比为1:0.05~0.5,碱与邻卤苯甲酰胺的摩尔比为1:0.5~3.5。
优选地,所述氰氨与邻卤苯甲酰胺的摩尔比为1:1.0~20,铜盐与邻卤苯甲酰胺的摩尔比为1:0.05~0.5,碱与邻卤苯甲酰胺的摩尔比为1:1.5~3.0。
优选地,所述反应在溶剂存在下进行,所述溶剂为水、甲醇、乙醇、乙腈、苯、甲苯、四氢呋喃、1,4-二氧六环、二甲基亚砜、N,N-二甲基甲酰胺、乙二醇、聚乙二醇(PEG-200~600)中的一种或多种;
所述铜盐包括但不仅限于氯化亚铜、溴化亚铜、碘化亚铜、氯化铜、溴化铜、碘化铜、碱式碳酸铜、碳酸铜、醋酸铜、甲酸铜、苯甲酸铜、三氟甲磺酸铜、三氟甲磺酸亚铜中的一种或多种;
所述碱包括但不仅限于氢氧化钠、氢氧化钾、氢氧化铯、碳酸钠、碳酸钾、碳酸铯、碳酸氢钠、碳酸氢钾、甲酸钠、甲酸钾、乙酸钠、乙酸钾、叔丁醇钠、叔丁醇钾、磷酸钾、DBU中的一种或多种。
优选地,所述反应温度为60~140℃,反应时间为1~24h。
优选地,所述反应温度为80~130℃,反应时间为4~12h。
优选地,所述反应在空气或惰性气氛下进行,所述惰性气氛为氮气气氛或氩气气氛。
优选地,所述氰氨为氰氨化钙、氰氨化锂、氰胺化钠、氰胺化钾中的一种,加入水做助催化剂,水与邻卤苯甲酰胺的摩尔比为1:0.5~3。
优选地,所述反应结束后,将反应液采用乙酸乙酯进行萃取,利用有机相水洗多次并使用无水硫酸镁干燥,最后有机相浓缩得到胺基喹唑啉酮衍生物。
优选地,所述浓缩采用常压蒸馏、减压蒸馏、旋转蒸发中的一种。
优选地,也可以利用柱层析纯化进行后处理,所述柱层析以200~300目硅胶为分离树脂,洗脱剂选择石油醚、正己烷、二氯甲烷、水、乙腈、甲醇、乙酸乙酯中的至少一种。
优选地,所述喹唑啉酮类衍生物结构式如下所示:
本发明实施例提供的一种胺基喹唑啉酮衍生物的制备方法,铜盐催化剂通过配合方式引入到邻卤苯甲酰胺中促进碳氮键的形成,并在碱的作用下脱离得到喹唑啉酮衍生物,原料、催化剂更易得,可有效降低生产成本、提高收率和化学选择性,可以便捷地应用于制备各喹唑啉酮衍生物。
附图说明
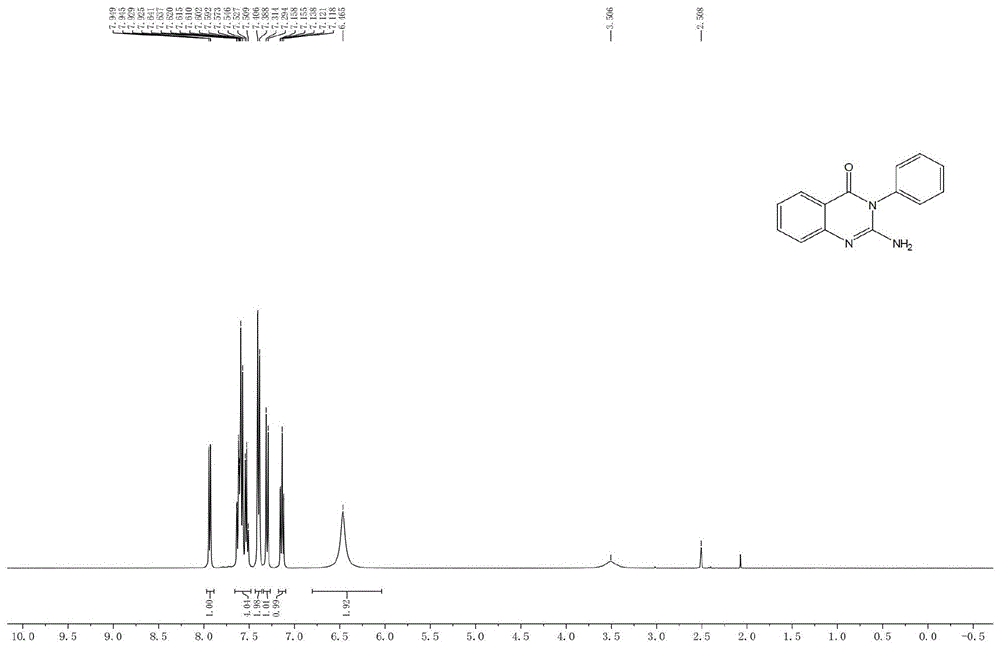
图1为本发明实施例1提供的化合物5a的核磁1H NMR谱图;
图2为本发明实施例1制备的化合物5a的核磁13C NMR碳谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
以下结合具体实施例对本发明的具体实现进行详细描述。
实施例1
一种胺基喹唑啉酮衍生物(化合物5a)的制备方法,其结构和制备方法如下:
方法一:
具体步骤为:称取单氰胺(1.5mmol,63mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰苯胺(1mmol,275mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),将其移入110℃的油浴锅中并搅拌,反应过夜,用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温,向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-苯基喹唑啉-4(3H)-酮,白色固体,分离收率83.2%。
方法二:
具体步骤为:称取石灰氮(1.8mmol,148mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰苯胺(1mmol,275mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),加入水(3.6mmol,65mg),将其移入110℃的油浴锅中并搅拌,反应过夜,用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温。向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-苯基喹唑啉-4(3H)-酮,白色固体,分离收率46.5%。
化合物5a的结构鉴定:
核磁共振数据:
1
13
化合物5a的
实施例2
一种胺基喹唑啉酮衍生物(化合物5b)的制备方法,其结构和制备方法如下:
具体步骤为:称取单氰胺(1.5mmol,63mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰对甲氧基苯胺(1mmol,305mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),将其移入110℃的油浴锅中并搅拌,反应过夜。用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温,向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-对甲氧基苯基喹唑啉-4(3H)-酮,白色固体,分离收率88.4%。
化合物5b的结构鉴定:
核磁共振数据:
1
13
化合物5b的
实施例3
一种胺基喹唑啉酮衍生物(化合物5c)的制备方法,其结构和制备方法如下:
具体步骤为:称取单氰胺(1.5mmol,63mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰对甲基苯胺(1mmol,299mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),将其移入110℃的油浴锅中并搅拌,反应过夜。用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温,向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-对甲基苯基喹唑啉-4(3H)-酮,白色固体,分离收率87.3%。
化合物5c的结构鉴定:
核磁共振数据:
1
13
化合物5c的
实施例4
一种胺基喹唑啉酮衍生物(化合物5k)的制备方法,其结构和制备方法如下:
具体步骤为:称取单氰胺(1.5mmol,63mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰对三氟甲氧基苯胺(1mmol,416mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),将其移入110℃的油浴锅中并搅拌,反应过夜。用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温,向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-对三氟甲氧基苯基喹唑啉-4(3H)-酮,白色固体,分离收率76.2%。
化合物5k的结构鉴定:
核磁共振数据:
1
13
化合物5k的
实施例5
一种胺基喹唑啉酮衍生物(化合物5u)的制备方法,其结构和制备方法如下:
具体步骤如下:称取单氰胺(1.5mmol,63mg),叔丁醇钾(2mmol,224mg),邻溴苯甲酰间二甲基苯胺(1mmol,304mg),碘化亚铜(0.1mmol,19mg)于25mL圆底烧瓶中,加入磁子,塞好橡胶塞,用高纯氮气置换三次后,在氮气保护下向烧瓶中加入DMSO(2mL),将其移入110℃的油浴锅中并搅拌,反应过夜。用TLC检测跟踪反应,反应结束后,将烧瓶冷却至室温,向体系中加入10mL饱和食盐水,搅拌;用乙酸乙酯萃取(10mL×3),合并有机相,用旋转蒸发仪去除溶剂,得到粗产品;粗产品经过硅胶负载,洗脱剂采用体积比石油醚:乙酸乙酯=1:1进行柱层析纯化后,得纯品2-氨基-3-(3,5-二甲基)苯基喹唑啉-4(3H)-硫酮,白色固体,分离收率76.1%。
化合物5u的结构鉴定:
核磁共振数据:
1
13
化合物5u的
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 喹唑啉-2,4-二酮衍生物及其制备方法和用途
- 哒嗪6,1-b并喹唑啉酮衍生物及其制备方法和应用
- 一种二氢喹唑啉酮和喹唑啉酮衍生物的磺酸类离子液体催化调控合成方法
- 异噁唑甲酰胺基-4(3H)-喹唑啉酮衍生物及其合成方法和用途